Вы точно человек?
Содержание:
Факторы апоптоза
Структурно-морфологические и биохимические изменения при апоптозе осуществляются определенным набором специализированных клеточных инструментов, среди которых наиболее важными являются каспасы, нуклеазы и мембранные модификаторы.
Каспазы – группа ферментов, разрезающих пептидные связи по остаткам аспарагина, фрагментируя белки на крупные пептиды. До начала апоптоза присутствуют в клетке в неактивном состоянии из-за ингибиторов. Главной мишенью каспаз являются ядерные белки.
Нуклеазы – ответственны за разрезание молекул ДНК. Особо важна в развитии апоптоза активная эндонуклеаза CAD, разрывающая участки хроматина в областях линкерных последовательностей. В результате образуются фрагменты длиной 120-180 нуклеотидных пар. Комплексное воздействие протеолитических каспаз и нуклеаз приводит к деформации и фрагментации ядра.
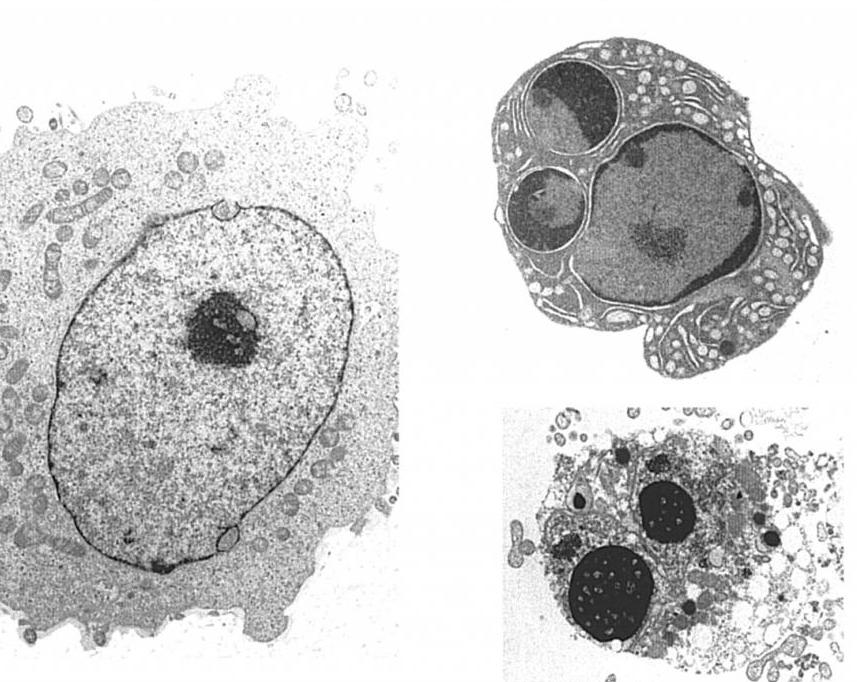
Модификаторы клеточной мембраны – нарушают асимметричность билипидного слоя, превращая его в мишень для фагоцитирующих клеток.
Ключевая роль в развитии апоптоза принадлежит каспазам, которые поэтапно активируют все последующие механизмы деградации и морфологической перестройки.
Инструменты апофоза
Одним из важнейших инструментов апоптоза является специальное семейство цитоплазматических протеаз, или так называемых каспаз. По структуре они относятся к сериновым протеазам по наличию в активном центре остатка — аминокислоты серина.
В своих белковых мишенях каспазы разрывают пептидные связи, образованные с участием остатка аспарагиновой кислоты. Всего в семействе каспаз 10 ферментов, поэтому каспазы способны в определенной последовательности активировать друг друга, образуя своего рода «каскад», причем разветвленный. С учетом этой разветвленной структуры каспаз, не вдаваясь в подробности ее структуры, которая достаточно сложна и во многом еще не выяснена, можно видеть, что главная их функция состоит в избирательном и ограниченном протеолизе определенных белковых мишеней (в основном ядерных белков), без затраты значительных количеств энергии.
В хорошо настроенном «оркестре» апоптоза (во многом благодаря предсуществующей генетической запрограммированности феномена на самоуничтожение клетки), в котором роль «первой скрипки» исполняет семейство цитоплазматических протеаз — каспаз, слажено «звучат» и другие факторы, такие как, например, эндонуклеазы, которые рассматриваются вторым по важности (после каспаз) «инструментом» апоптоза. Не вдаваясь в детали этой довольно сложной и во многом еще не понятной последовательности событий, здесь для нашей задачи целостного рассмотрения феноменов клеточной пролиферации и апоптоза следует указать на преимущественно ядерную локализацию эндонуклеаз
Не вдаваясь в детали этой довольно сложной и во многом еще не понятной последовательности событий, здесь для нашей задачи целостного рассмотрения феноменов клеточной пролиферации и апоптоза следует указать на преимущественно ядерную локализацию эндонуклеаз.
Эти белки-ферменты обладают двумя важными свойствами. Во-первых, эндонуклеазы в отличие от других ферментов, осуществляющих аутолиз ДНК, расщепляют ДНК хромосом не в случайных местах, а только в линкерных участках. Поэтому хроматин не подвергается полному лизису, а лишь фрагментируется, что составляет отличительную черту апоптоза.
И наконец, последнее, но не менее важное. Речь идет о белке р53, который рассматривается как универсальный фактор, тормозящий клеточную пролиферацию, а при определенных условиях и опухолевую трансформацию
В ходе многих, если не всех видов апоптоза в клетках возрастают содержание и активность белка р53. Дело в том, что белок р53 стимулирует гены ряда «киллерных» рецепторов, т. е. рецепторов, воспринимающих «команду» начала апоптоза. Среди них белки Fas-рецептор и рецептор KILLER/DR5. Таким путем повышается чувствительность клетки к сигналам, запускающим апоптоз.
Другой важной функцией белка р53 является его способность останавливать клеточный цикл. Это происходит благодаря активации гена Р21, продукт которого (белок р21) ингибирует известные уже нам комплексы циклинов (циклин-Cdks)
Очень важно иметь в виду воздействие белка р53 и на окружающие клетки. Белок р53 активирует и такие гены, продукты которых выделяются из гибнущей клетки и воздействуют на ее окружение, например подавление ангиогенеза (торможение опухолевого роста), и, что особенно важно в свете рассматриваемой проблемы, р53 действует как фактор, стимулирующий синтез и секрецию ряда ингибиторов пролиферации клеток
Даже из далеко не полного списка функций белка р53 видно, что этот фактор исполняет роль «первой скрипки» в слаженном звучании оркестра «ферментов-исполнителей» и внимательно следит за «хромосомным дирижером», с тем чтобы не пропустить такт своего вступления, или наоборот, выдержать необходимую паузу.
По материалам: health-medicine.info
Терапия на основе апоптоза.
Таким образом, апоптоз является общебиологическим механизмом, ответственным за поддержание постоянства численности клеточных популяций, а также формообразование и выбраковку дефектных клеток. Нарушение регуляции апоптоза приводит к возникновению различных заболеваний, связанных с усилением или, наоборот, ингибированием апоптоза. Следовательно, изучение механизмов регуляции различных этапов данного процесса позволит определенным образом воздействовать на его отдельные этапы с целью их регуляции или коррекции. В настоящее время общепринято: если клетка погибает от апоптоза — подразумевается возможность терапевтического вмешательства, если вследствие некроза — нет. На основе знаний о программированной гибели клетки используется широкий ряд препаратов с целью регуляции этого процесса в различных типах клеток. Так, сведения о рецептор-опосредованной регуляции апоптоза клеток позволяют использовать их для терапии гормон-зависимых новообразований. С использованием андроген-блокирующей терапии лечат рак простаты. Рак молочной железы часто подвергается регрессии при применении антагонистов эстрогеновых рецепторов. Информация о биохимических сигнал-передающих путях регуляции апоптоза позволяет эффективно применять антиоксидантную терапию, а таюке использовать препараты, регулирующие концентрацию кальция, либо активирующие (ингибирующие) различные протеинкиназы, с целью коррекции апоптоза в различных типах клеток. Осознание роли апоптоза в гибели клеток интенсифицировало поиск фармакологических средств, защищающих их от апоптоза. Активно изучаются ингибиторы специфических протеаз в качестве фармакологических агентов. Это, как правило, три- или тетрапептиды, содержащие аспарагин. Ограничением их терапевтического использования является их низкая способность проникать в клетку. Однако, несмотря на это, в экспериментах in vivo получены успешные результаты при использовании N-бензилокси-карбонил-Vа1-А1а-Аsр-фторметилкетона ингибитора различных каспаз для снижения зоны инфаркта при моделировании инсульта. В ближайшие годы можно ожидать появления новых лекарственных препаратов для лечения и предупреждения различных заболеваний, в основе действия которых будет заложен принцип регуляции процессов апоптоза.
Многообещающими являются также подходы, связанные с регуляцией апоптоз-специфических генов и реализующиеся, в частности, в генной терапии — одной из самых перспективных областей современной медицины — при лечении заболеваний, вызванных нарушением функционирования отдельных генов. Идентификация морфологических и биохимических маркеров апоптоза должна в перспективе способствовать более глубокому пониманию механизмов патогенеза заболеваний, улучшению дифференциальной диагностики и созданию принципиально новых направлений терапии.
Апоптоз дефинитивных клеток
Отдельные клетки, как и организм в целом, подвергаются старению. Такие клетки называют дефинитивными клетками, или клетками в состоянии терминальной (конечной) дифференцировки. Каждый дифферон имеет свои дефинитивные клетки. Например, фиброциты — дефинитивные клетки фибробластического дифферона, гранулоциты крови и макрофаги — миеломоноцитарного дифферона, хондроциты и остеоциты — дефинитивные клетки соответственно хондро- и остеобластического дифферонов и т.п. Клетки, находящиеся в состоянии терминальной дифференцировки разрушаются, как правило, путём апоптоза.
Апоптоз эмбриогенеза
Часть клеток эмбриона в ходе органогенеза погибает путём апоптоза. Так, например, формируются межпальцевые промежутки. Кроме того, в теле зародыша происходят интенсивные клеточные перемещения, сопровождающиеся неизбежными ошибками в виде избыточного накопления клеток в одних участках эмбриона, недостаточной их продукции в других или появления клеток с нехарактерным направлением дифференцировки. Избыточная клеточная масса также разрушается путём апоптоза.
Особенности апоптоза эмбриогенеза:
1. Часто (но не всегда) при апоптозе эмбриональных клеток процесс сопровождается аутофагией, которая не развивается при апоптозе клеток в более поздние сроки онтогенеза. При этом происходит активация лизосом с последующей ферментативной деградацией разрушающейся клетки или апоптозных телец. Из органелл наиболее быстро деструкции подвергаются митохондрии. Однако несмотря на процессы аутофагоцитоза плазмалемма в апоптозных тельцах длительно остаётся неповреждённой.
2. Фагоцитоз апоптозных телец в организме эмбриона более продолжителен (несколько часов) в отличие от тканей взрослого организма, где апоптозные тельца обычно фагоцитируются в течение нескольких минут. Однако длительность фазы деградации апоптозных телец нельзя считать особенностью апоптоза эмбриогенеза; скорее она является особенностью функции фагоцитарных клеток в раннем онтогенезе.
По механизму апоптоз эмбриогенеза протекает как «отмирание по умолчанию». Гибель части клеток происходит вследствие недостаточного содержания в межклеточном матриксе морфогенетических молекул, являющихся факторами выживания для эмбриональных клеток. Для каждого типа клеток зародыша существуют определённые морфогенетические факторы, от концентрации которых зависит общая масса той или иной клеточной популяции.
Патология апоптоза эмбриогенеза
Различают две формы патологии апоптоза эмбриогенеза: чрезмерно выраженный апоптоз эмбриогенеза и его недостаточность.
1. Чрезмерновыраженный апоптоз эмбриогенеза сопровождается дефицитом клеточной массы в тех или иных участках тела зародыша. Это может привести к формированию пороков развития в виде агенезии (полное отсутствие органа), аплазии (отсутствие органа при сохранении эмбрионального зачатка), врождённой гипоплазии (недоразвитие органа), атрезии (полное отсутствие канала или естественного отверстия) и врождённого стеноза (сужение канала или отверстия), а также тканевыми пороками, не всегда проявляющимися клинически, но создающими основу для возникновения различной приобретённой патологии в постнатальном онтогенезе.
2. Недостаточность апоптоза эмбриогенеза ведёт к возникновению двух типов тканевых пороков развития — гамартий и хористий.
Гамартии (от греч. ἁμαρτία ἡ = ἁμάρτημα, τος τό — ошибка, заблуждение, преступление; лат. hamartia) — избыточно развитый нормальный компонент ткани. Например, гемангиомы и пигментные невусы, хондромы (гамартохондромы) лёгкого являются типичными гамартиями.
Хористии (от греч. χωρίζω — отделять, отличать; χωρίς — отдельно) — появление в органе нехарактерных для него тканевых структур. Примерами наиболее распространённых хористий являются дермоидные кисты различной локализации и струма (зоб) яичника. В качестве синонима хористии нередко используется термин «гетеротопия», однако им обозначают не только тканевые изменения, сформировавшиеся во внутриутробном периоде, но и приобретённые в постнатальном онтогенезе, например, гетеротопия эндоцервикального эпителия на вагинальной порции шейки матки (эндоцервикоз).
НЕКРОЗ
Некроз — собственно смерть повреждённой клетки, сопровождающаяся необратимым прекращением её жизнедеятельности. Некроз является завершающим этапом клеточных дистрофий или следствием прямого действия на клетку повреждающих факторов значительной (разрушающей) силы. Некроз, как правило, сопровождается воспалительной реакцией.
Паранекроз и некробиоз.
Некрозу предшествуют паранекроз (метаболические и структурные изменения ещё обратимы) и некробиоз. На этапе некробиоза патогенные изменения приобретают необратимый характер и приводят к некрозу. Основные звенья патогенеза некроза те же, что и при повреждении клеток, но при развитии некроза они максимально интенсифицированы и развиваются на фоне недостаточности адаптивных механизмов (защиты и регенерации повреждённых структур, компенсации нарушенных процессов в клетке).
Лизис и аутолиз.
Некротизированные клетки подвергаются деструкции (лизис) при помощи лизосомных ферментов и свободных радикалов.
- Гидролиз внутриклеточных компонентов и межклеточного вещества происходит под влиянием ферментов лизосом альтерированных клеток. Высвобождению лизосомных ферментов способствует развитие внутриклеточного ацидоза.
- Деструкция повреждённых компонентов клеток осуществляется при участии активных форм кислорода и свободных радикалов. Известны факты интенсификации свободнорадикальных и липопероксидных реакций при остром воспалении, механическом повреждении, на определённых этапах инфаркта (частной формы некроза, развивающегося вследствие нарушения кровоснабжения ткани), опухолевого роста (сопровождается гибелью большого числа как злокачественных, так и окружающих нормальных клеток) и других патологических процессах.
Эти два механизма обеспечивают саморазрушение структур клетки (аутолиз).
Разрушение повреждённых и некротизированных клеток происходит и при участии других клеток — фагоцитов, а также микроорганизмов. В отличие от аутолитического распада, последний механизм обозначают как гетеролитический.
АПОПТОЗ
Апоптоз является другим вариантом гибели клеток.
АПОПТОЗ — форма гибели отдельных клеток, возникающая под действием вне- или внутриклеточных факторов, осуществляющаяся путём активации специализированных внутриклеточных процессов, регулируемых определёнными генами.
Таким образом, апоптоз — программированная гибель клетки. В этом его принципиальное отличие от некроза. Другое принципиальное отличие апоптоза от некроза состоит в том, что программу апоптоза запускает информационный сигнал, тогда как некроз клетки развивается под влиянием повреждающего агента. В финале некроза происходит лизис клетки и освобождение её содержимого в межклеточное пространство, тогда как апоптоз завершается фагоцитозом фрагментов разрушенной клетки. Некроз — всегда патология, тогда как апоптоз наблюдается в ходе многих естественных процессов, а также при адаптации клетки к повреждающим факторам. Апоптоз — в отличие от некроза — энергозависим и требует синтеза РНК и белков.
Примеры апоптоза.
Запрограммированная гибель клеток; Гибедь клеток, выполнивших свою функцию; Дегенерация; Ликвидация аутоагрессивных Т-клеток; Старение; Трансфекция; Повреждение клетки; Опухолевый рост.
Механизм апоптоза.
При реализации апоптоза условно можно выделить четыре стадии.
Стадия инициации.
На этой стадии информационные сигналы рецептируются клеткой. Патогенный агент либо сам является сигналом, либо обусловливает генерацию сигнала в клетке и его проведение к внутриклеточным регулятор-ным структурам и молекулам.
Стадия программирования.
На этой стадии специализированные белки либо реализуют сигнал к апоп-тозу путём активации исполнительной программы (её эффекторами являются цистеиновые протеазы — каспазы и эндонуклеазы), либо блокируют потенциально летальный сигнал.
Стадия реализации программы.
Стадия реализации программы апоптоза (исполнительная, эффекторная) состоит в собственно гибели клетки, осуществляемой посредством активации протеолитического и нуклеолитического каскадов.
Стадия удаления фрагментов погибших клеток.
На поверхности апоптозных телец экспрессируются лиганды, с которыми взаимодействуют рецепторы фагоцитирующих клеток. Фагоциты быстро обнаруживают, поглощают и разрушают апоптозные тельца. Благодаря этому содержимое разрушенной клетки не попадает в межклеточное пространство, а при апоптозе отсутствует воспалительная реакция. Этот признак отличает апоптоз от некроза, который сопровождается развитием перинекротического воспаления.
Механизмы индукции апоптоза при повреждении ДНК.
До последнего времени считалось, что нерепарируемые повреждения ДНК приводят клетку к гибели в результате нарушения функций всех биохимических систем из-за невозможности полноценной транскрипции генов, содержащих дефекты в матрице ДНК. Исследования последних лет привели к формированию принципиально новых представлений о механизме гибели клеток, имеющих повреждения ДНК, как о процессе, осуществляемом в соответствии с определенной генетической программой. В индукции этой программы при наличии повреждений в ДНК клетки важная роль принадлежит белку р53. Этот белок с молекулярной массой 53 кДа локализован в ядре клетки и является одним из транскрипционных факторов, повышенная экспрессия которого приводит к репрессии ряда генов, регулирующих транскрипцию и причастных к задержке клеток в фазе клеточного цикла G1. При повреждении ДНК под действием ионизирующего излучения или УФ-излучения, ингибиторов топоизомеразы II и некоторых других воздействиях происходит активация экспрессии гена р53. Блок клеточного цикла в фазах G1 и G2 до репликации ДНК и митоза, соответственно, делает возможной репарацию поврежденной ДНК и предотвращает тем самым появление мутантных клеток. Если же активность репарационных систем недостаточна и повреждения ДНК сохраняются, то в таких клетках индуцируется программируемая клеточная гибель, или апоптоз, что приводит к защите организма от присутствия клеток с поврежденной ДНК, т.е. мутантных и способных к злокачественной трансформации.
На уровне транскрипции р53 регулирует экспрессию генов, участвующих в блокаде клеточного цикла — р21 (ингибитор большинства циклин-зависимых киназ, либо взаимодействует либо с комплексами, определяющими синтез и репарацию ДНК, либо с белками, модулирующими апоптоз — Вах. Последовательность рассмотренных событий представлена на рис. 7.
Мутации гена р53 позволяют таким клеткам сохранять жизнеспособность в митозе, что чревато выживанием клеток, подвергшихся опухолевой трансформации. И действительно, при онкологической трансформации обнаружено значительное количество мутаций гена р53. Мутации гена р53 связаны с плохим прогнозом в лечении злокачественных новообразований. Такие опухолевые клетки оказываются резистентными к лучевой и химиотерапии. И, наоборот, опухоли с нормальным (диким типом) р53 — легко поддаются лечению.
Таким образом, при действии генотоксических агентов р53 не только увеличивает время репарации ДНК. но также защищает организм от клеток с опасными мутациями. Блокирование процесса апоптоза, происходящее на разных стадиях канцерогенеза, приводит к снижению способности трансформированных клеток активировать программу клеточной гибели, что определяет прогрессию опухолевого заболевания.
Роль белков семейства Вс1-2 в регуляции апоптоза клетки.
Процесс регулированной клеточной гибели условно может быть разделен на несколько различных фаз: фаза инициации апоптоза, проведение сигнала, активация каспаз, активация эндонуклеаз и специфическая деградация ДНК, в результате чего наступает гибель клетки.
Если начальные фазы различаются в зависимости от типа клеток и от апотоз-индуцирующего сигнала, то этап деградации ДНК — универсален для большинства клеток. Эта фаза является переходом к необратимой — терминальной стадии апоптоза, которую контролируют белки семейства Вс1-2, производные одноименных генов (рис. 8). В связи с этим, выяснение роли белков семейства Вс1-2 занимает центральное место в изучении регуляции процесса апоптоза. К настоящему времени известно, что белки этого семейства относятся либо к индукторам апоптоза (Bad, Bax, Bcl-Xs, Bik, Bid, Bak), либо к ингибиторам (Bcl-2, Bcl-XL). Белки семейства Bcl-2 находятся в постоянном динамическом равновесии, образуя гомо- и гетеродимеры, что в конечном счете влияет на развитие апоптоза клеток. Поэтому считается, что соотношение активных форм этих белков определяют реостат жизни и смерти клетки. Механизм регуляции этого процесса целесообразно рассматривать с позиции структурно-функциональных взаимоотношений между белками этого семейства, которые позволяют объединить их в одно семейство — белков семейства Bcl-2.
Реакция клеток на внешнее воздействие.
Описанная морфология клеток не является стабильной (постоянной). При воздействии на организм различных неблагоприятных факторов в строении различных структур проявляются различные изменения. В зависимости от факторов воздействия изменения клеточных структур проявляются неодинаково в клетках разных органов и тканей. При этом изменения клеточных структур могут быть адаптивными (приспособительными) и обратимыми, или жедезадаптивными, необратимыми (патологическими). Однако определить четкую грань между адаптивными и дезадаптивными изменениями не всегда возможно, так как приспособительные изменения могут перейти в патологические. Поскольку объектом изучения гистологии являются клетки, ткани и органы здорового организма человека, то здесь будут рассмотрены прежде всего адаптивные изменения клеточных структур. Изменения отмечаются как в строении ядра, так и цитоплазмы.
Изменения в ядре— набухание ядра и сдвиг его на периферию клетки, расширение перинуклеарного пространства, образование инвагинаций кариолеммы (впячивание внутрь ядра его оболочки), конденсация хроматина. Кпатологическим изменениям ядра относят:
-
пикноз — сморщивание ядра и коагуляция (уплотнение) хроматина;
-
кариорексис — распад ядра на фрагменты;
-
кариолизис — растворение ядра.
Изменения в цитоплазме— уплотнение, а затем набухание митохондрий, дегрануляция зернистой эндоплазматической сети (слущивание рибосом), а затем и фрагментация канальцев на отдельные вакуоли, расширение цистерн, а затем распад на вакуоли пластинчатого комплекса Гольджи, набухание лизосом и активация их гидролаз, увеличение числа аутофагосом, в процессе митоза — распад веретена деления и развитие патологических митозов.
Изменения цитоплазмымогут быть обусловлены структурными изменениями плазмолеммы, что приводит к усилению ее проницаемости и гидратации гиалоплазмы, нарушением обмена веществ, что сопровождается снижением содержания АТФ, снижением расщепления или увеличением синтеза включений (гликогена, липидов) и их избыточном накоплении.
После устранения неблагоприятных воздействий на организм реактивные(адаптивные) изменения структур исчезают и морфология клетки восстанавливается. При развитиипатологических(дезадаптивных) изменений даже после устранения неблагоприятных воздействий структурные изменения нарастают и клетка погибает.