Болезнь вильсона-коновалова
Содержание:
- Перед визитом к врачу
- Как лечат болезнь Вильсона беременным женщинам?
- Лечение
- Формы и симптомы
- Патогенез
- Клиническая картина болезни Вильсона
- История[править | править код]
- Общая картина заболевания
- Осложнения и последствия
- Симптомы патологии
- Терапия патологии
- Диета
- Генетика
- Каким образом наследуется болезнь?
- Диагностические меры
Перед визитом к врачу
Для постановки точного диагноза и назначения терапии нужно обратиться к специалисту по заболеваниям печени – гепатологу. Однако в том случае, если вы впервые обращаетесь в медицинское учреждение с жалобами на симптомы, схожие с признаками болезни Вильсона, следует начать с посещения терапевта. При необходимости он перенаправит вас к нужному специалисту.

Так как медицинские консультации чаще всего ограничены во времени, а врачи при этом стремятся донести до пациентов максимум материала, лучше подготовиться к визиту заблаговременно. Специалисты дают следующие рекомендации:
Уточните, существуют ли условия и ограничения, которых необходимо придерживаться перед визитом к врачу. В некоторых случаях требуется соблюдать определенную диету или заранее сдать анализы крови.
Составьте подробный перечень всех симптомов, которые, по вашему мнению, свидетельствуют о наличии патологии
Важно записать даже признаки, на первый взгляд не относящиеся к проблеме.
Запишите ключевые личные данные, в том числе недавние события, повлекшие сильную психоэмоциональную перегрузку (стресс), изменения в образе жизни, обнаружение болезни Вильсона в семейном анамнезе.
Составьте список всех лекарственных препаратов, витаминов и других регулярно принимаемых пищевых добавок.
Возьмите с собой близкого родственника или хорошего друга. В некоторых случаях тяжело сразу воспринять весь объем информации, предлагаемой врачом, а друг или член семьи способен запомнить или письменно зафиксировать детали, которые могут ускользнуть от вашего внимания.
Как лечат болезнь Вильсона беременным женщинам?
Беременные женщины должны продолжать лечение болезни на протяжении всей беременности. Врачи назначат более низкие дозы хелатирующих препаратов. Поскольку плод нуждается в небольшом количестве меди, снижение дозы может удерживать медь на безопасном уровне без удаления слишком большого количества.
В большинстве случаев врачи рекомендуют женщинам продолжать принимать полную дозу цинка во время беременности. Эксперты рекомендуют женщинам с болезнью Вильсона не кормить грудью, если принимают хелатирующие препараты. Пеницилламин присутствует в грудном молоке и может быть вредным для ребенка. О безопасности триентина и цинка, содержащегося в грудном молоке имеется мало информации.
Лечение
Схема лечения Вильсона-Коновалова подбирается в индивидуальном порядке. Основная задача медикаментозной терапии заключается в выведении избыточного количества меди из организма пациента.

Программа лечения от болезни Вильсона-Коновалова состоит из следующих этапов:
- Диагностика со сбором требуемых анализов и тестов;
- Получение консультации у специалистов с назначением индивидуальной схемы лечения;
- Осуществление медикаментозных и лечебных процедур для устранения симптомов заболевания;
- Прохождение реабилитационных мер в отделении неврологии.
Основные методы лечения Вильсона-Коновалова:
- Прием Д-пенициламина, уменьшающего количество меди в кровяных тельцах больного. Данный вариант лечения относится к патогенетическому;
- Реабилитационные меры в отделении неврологии для устранения негативного воздействия на нервную систему больного;
- Внутренний прием сорбентов и гепатопротекторов (при наличии повреждений печеночных тканей);
- Трансплантация печени при наличии серьезных, не возобновляемых повреждений органа;
- С помощью специализированного оборудования осуществляются процедуры по очистке плазмы крови от токсинов и свободной меди (гемоиммуносорбция), удалению из плазмы продуктов патологического распада (мембранный плазмаферез). Пациенту могут быть назначены до 3 процедур п очистке крови. Данная методика лечения зарекомендовала себя, как максимально эффективная.

Первые результаты от очистки крови заметны уже после 14 календарных дней. Медикаментозное лечение может длиться до полугода.
Кроме того, любая схема лечения от болезни Вильсона-Коновалова сопровождается приемом медикаментов. Среди них чаще всего выписывают:
Лекарственные препараты, снижающие количество меди в организме больного (хелаты);
Медикаменты, способствующие регенерации печеночных клеток;
Желчегонные лекарственные медикаменты;
Комплекс витаминов (важно применение В6);
Лекарства, блокирующие поступление меди в организм с продуктами питания (например, содержащие цинк);
Медикаменты с антиоксидантными свойствами, снижающие интоксикацию организма;
Средства, подавляющие действие иммунной системы;
Лекарственные препараты успокоительного характера для устранения неврологических расстройств;
Средства, обладающие противовоспалительным результатом.
Назначенные доктором медикаменты пациент обязан принимать с начала диагностирования болезни и до конца его жизни. Если больной снизит дозировку или полностью прекратит лечение, то Вильсон-Коновалов может вернуться с более сильной симптоматикой.
В запущенных случаях заболевания, когда медикаментозные методы не дают должного результата, а состояние больного ухудшается диагностирована печеночная недостаточность, назначается трансплантация печени. Новая печень приживается и нормально функционирует уже через месяц после операции.

Формы и симптомы
Существует несколько форм болезни, симптомы которой проявляются после 20 или 40 лет.
На начале развития болезни у пациентов появляются только психические нарушения, лихорадка, поражение суставов или анемия. При переизбытке меди возможно развитие сахарного диабета, аневризмы (изменения сосудов) и синдром Фанкони (нарушение обмена веществ). Для болезни характерен клинический полиморфизм, т. е. первые симптомы патологии могут возникнуть в детстве, юношестве и в зрелом возрасте.
К другим осложнениям болезни Вильсона-Коновалова относятся:
- фульминантный гепатит (отмечается массовое разрушение клеток печени, ее уменьшение в размерах и летальный исход);
- образование камней в почках.
Печеночная форма
Встречается в 50-80% случаев. Формируется с 11-летнего возраста. Отмечается развитие хронического гепатита или цирроза печени. У больных происходит окрашивание кожи и слизистых в желтый цвет (желтуха). Патология появляется из-за увеличения количества билирубина в крови. Сначала происходит пожелтение склер и других слизистых оболочек. В первую очередь желтеет кожа лица, а потом верхние и нижние конечности. Область живота меняет свой цвет последней. При этом заболевании у пациента появляется печеночная форма желтухи, происходит поражение клеток печени, а цвет кожи становится желто-оранжевым.
Иногда пациенты жалуются на зуд. Моча становится темной, а кал — светлым. При обследовании у больных можно обнаружить асцит (скопление жидкости в брюшной полости), что характеризуется увеличением размеров живота. У пациентов наблюдаются отеки под глазами и кровотечения. Отмечается появление Кольца Кайзера-Флейшера (на наружном крае роговицы глаза можно обнаружить кольцо коричневого цвета, которое формируется из-за скопления меди). Особенностью болезни у женщин является аменорея — отсутствие менструальных циклов.
Неврологическая форма
Происходит поражение мозжечка, коры головного мозга и базальных ганглиев. Развивается патология после 19 лет. У пациентов наблюдается тремор (непроизвольные быстрые движения) рук и головы, который может быть постоянным или возникать периодически. Эта форма патологии часто сопровождается гримасничаньем, т. е. происходят быстрые изменения движения мышц лица. Наблюдаются изменения почерка: он становится неразборчивым и неровным, а буквы — неодинаковыми по размеру и расположению в слове. У пациентов происходят нарушения в произношении звуков и слов (дизартрия).
Отмечается контрактура (ограничение движений в суставах) и повышенный тонус мышц. У 20% больных встречаются нарушения психики (депрессия, которая сопровождается снижением настроения, двигательной заторможенностью и апатией; психоз, характеризующийся появлением галлюцинаций и бреда), иногда эпилептические припадки. Возможно возникновение Кольца Кайзера-Флейшера.
Редкие случаи
Более редкие формы патологии:
| Форма | Характеристика |
| Экстрапирамидно-корковая | Встречается редко. Отмечаются эпилептические припадки, дефицит интеллекта и двигательные нарушения |
| Ригидно-аритмогиперкинетическая | Дебют заболевания — детский возраст. Наблюдается мышечная ригидность (напряжение мышц), отсутствие мимики и смазанность речи, нарушение мелкой моторики. Снижение интеллекта происходит на умеренном уровне. Для этой формы болезни характерно прогрессирование с периодами обострения и ремиссий |
| Дрожательная | Возникает в возрасте 10-30 лет. Основным признаком является тремор. Встречаются эпилептические приступы и брадикинезия (снижение скорости движения при высоком тонусе мышц). Отмечается брадилалия (замедленная речь) и психоорганический синдром, который сопровождается снижением интеллектуальных способностей, ослаблением памяти и недержанием аффектов, т. е. нарушениями в эмоциональной сфере |
Редкие симптомы:
| Симптом | Характеристика |
| Изменения со стороны почек | Возникают в 10% случаев из-за накопления меди в клетках почек. Из-за это происходит их разрушение, у больных может развиться гематурия (присутствие в моче эритроцитов, которых в норме нет) и гликозурия (наличие глюкозы в моче) |
| Внутрисосудистый гемолиз | Характеризуется разрушением эритроцитов в полости сосудов. Появляется у 10-15% больных. Такая патология приводит к анемии (малокровию, которая заключается в снижении количества эритроцитов и гемоглобина в крови) |
| Поражение костной системы | Появляется в 20% случаев. При таком проявлении у пациентов возникает остеопороз — заболевание, при котором снижается плотность и нарушается структура костей. На этом фоне повышается ломкость костей, что приводит к постоянным переломам |
Патогенез
В основе патогенеза болезни Вильсона лежит нарушение выведения меди. Обычно в рационе содержится намного больше меди, чем необходимо организму. В норме избыток меди выводится с желчью, но при болезни Вильсона медь накапливается в печени.
На ранних стадиях болезни при гистологическом исследовании обнаруживают жировую дистрофию печени и отложения гликогена в ядрах гепатоцитов. При электронной микроскопии можно увидеть характерные изменения митохондрий. В дальнейшем развиваются некроз, воспаление, разрастание желчных протоков и цирроз печени. На всех стадиях повышается активность аминотрансфераз, изменяются и другие биохимические показатели функции печени. Со временем наступает перегрузка гепатоцитов медью, после чего медь поступает в кровь и захватывается другими тканями, прежде всего головным мозгом.
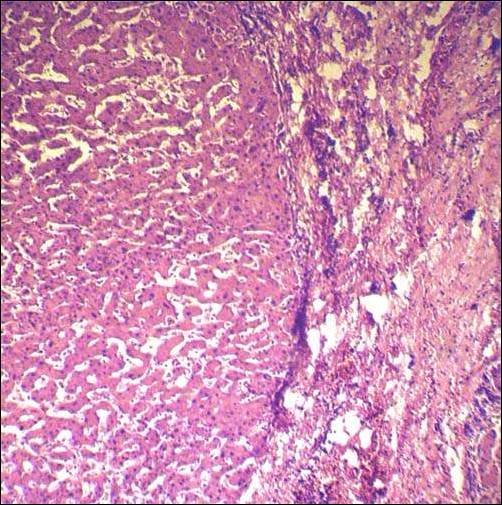 Гистологический препарат печени при болезни Вильсона (мальчик 6 лет, бессимптомная стадия). Видны крупно- и мелкокапельная жировая дистрофия печени, отложения гликогена в ядрах гепатоцитов и клеточная инфильтрация. Окраска гематоксилином и эозином.
Гистологический препарат печени при болезни Вильсона (мальчик 6 лет, бессимптомная стадия). Видны крупно- и мелкокапельная жировая дистрофия печени, отложения гликогена в ядрах гепатоцитов и клеточная инфильтрация. Окраска гематоксилином и эозином.
При МРТ обнаруживают признаки отложения меди в чечевицеобразных ядрах, реже — в варолиевом мосту, продолговатом мозге, таламусе, мозжечке и коре головного мозга. На ранних стадиях обнаруживают клетки Опальского и Альцгеймера II типа (не патогномоничны для болезни Вильсона), а позднее происходят гибель нейронов и образование кист в ткани мозга.
Отложение меди в почках почти не влияет на их структуру и функцию. Иногда наблюдаются микрогематурия или небольшая протеинурия, редко — нефрокальциноз, мочекаменная болезнь и почечный канальцевый ацидоз. Остальные органы и ткани при болезни Вильсона почти не страдают.
Клиническая картина болезни Вильсона
Еще одним распространенным названием заболевания является гепатолентикулярная дегенерация. При описании клинической картины обязательно указываются форма, выраженность нарушений со стороны нервной системы, уровень печеночной недостаточности. Код по МКБ 10 определяется как Е83.0.
Клинические проявления болезни возникают в раннем возрасте, они схожи с симптомами многих заболеваний печени. Большинство пациентов страдают от желтухи, астении, анорексии. У многих женщин отмечают постоянно повышенную температуру тела.
Печень больных насыщается медью, происходит накопление этого вещества во всем организме, в том числе нервной системе. Это отрицательно сказывается на мимике, моторике, координации движений. Интеллект при этом сохраняется, но поведение человека становится агрессивным.
Происходит превышение допустимого количества меди в роговице глаза, что сопровождается появлением на ней коричневого кольца. Его обнаруживают при помощи щелевой лампы, но только у пациентов старше 5 лет.
Синдром Вильсона — Коновалова характеризуется клиническим полиморфизмом, в процесс вовлекаются органы выделительной и нервной систем. Заболевание имеет рецессивные признаки, которым предшествуют висцеральные и желудочно-кишечные расстройства. В клинической картине описывают гепатолиенальный синдром, застой кровотока и мышечную ригидность без нарушения чувствительности.
История[править | править код]
Английский невролог Сэмюель Вильсон (англ. S. Wilson — более нормативная передача У́илсон) (1878 — 1937) в 1912 году описал типичные для гепато-церебральной дистонии изменения в головном мозге, установил постоянное наличие цирроза печени и дал описание клиники нового заболевания, названного им прогрессивной лентикулярной дегенерацией (лат. lenticularis чечевицеобразный).
В качестве основных симптомов заболевания были отмечены разнообразные непроизвольные движения конечностей и туловища, мышечная ригидность, приводящая к скованности, дисфагия и дизартрия, аффективные вспышки, иногда психические расстройства, но признаки поражения пирамидных путей отсутствовали. Ещё раньше К. Вестфалем () и А. Штрюмпелем () было описано заболевание, которое по клиническому сходству с рассеянным склерозом получило название «псевдосклероз». Заболевание характеризовалось распространёнными, размашистыми, ритмичными непроизвольными движениями, повышением мышечного тонуса, амимией, дизартрией и выраженными психическими нарушениями вплоть до такого расстройства интеллекта, как слабоумие.
В дальнейшем оказалось, что прогрессивная лентикулярная дегенерация и псевдосклероз являются разными формами одного и того же заболевания, которое Галль (1921) назвал гепато-лентикулярной дегенерацией. Однако изменения в мозге при нём никогда не ограничиваются лентикулярными ядрами и нередко бывают даже сильнее выражены в других отделах мозга.
В 1960 году советский невропатолог Н. В. Коновалов предложил название «гепато-церебральная дистрофия», значительно расширил представления о патофизиологии, патогенезе и клинике этой болезни и выделил 4 формы поражения нервной системы и одну абдоминальную.
Общая картина заболевания
Второе название данного заболевания – гепатоцеребральная дистрофия или «медный токсикоз». Впервые это заболевание было изучено в 1912 году британским ученым-медиком Вильсоном. Несколько позже его труды были переработаны и дополнены отечественным медиком Коноваловым. Отсюда и название болезни Вильсона-Коновалова.
Болезнь Вильсона в основном «атакует» молодых мужчин до 25 лет и подростков. Чаще всего заболевание проявляет симптоматику в возрасте 10-15 лет.
https://youtube.com/watch?v=CrHTZqS5QVs
Болезнь Вильсона-Коновалова имеет наследственный характер, то есть если у кого-то из родителей имело место быть такое заболевание, вероятность, что дети могут им заболеть, увеличивается в несколько раз.
Виновником данной болезни становится «сбой» в генотипе – один из генов, размещенных в 13-ой хромосоме, является дефектным. Именно он отвечает за нормальный обмен меди в организме. Если он «работает некорректно», в теле человека начинает вырабатываться и накапливаться избыточное количество металла (меди), которое постепенно отравляет весь организм. Медь откладывается в головном мозге и печеночных тканях.
Болезнь передается только от родителей к детям, но необязательно у кого-то из родителей будет присутствовать такое заболевание. Они могут быть носителями дефектного гена и передать его ребенку, у которого и проявится Вильсон-Коновалов. Когда один или оба родителя являются носителями, болезнь получит развитие только у ¼ младенцев.
Инфекционный характер заболевания полностью отсутствует, то есть контактирующие с больным люди не подвержены опасности.
Отмеченные случаи заболевания достаточное редкие – не более 3 больных на 100 000 здоровых человек.
Клиническая картина Вильсона-Коновалова такова, что:
- В организме начинается «сбой» процессов обмена меди, из-за чего начинается беспорядочная выработка меди, не выводящейся своевременно. Наблюдается избыток данного элемента;
- Повреждению сильнее всего подвержены: печень, головной мозг, органы зрения и почки;
- Снижается количество минеральных веществ в организме, что приводит к размягчению костных тканей и переломам.

Отсутствие лечения и своевременной диагностики приводит к летальному исходу. Обычно человек доживает до 30 лет. Смерть наступает в случае развития печеночной и почечной недостаточности.
Осложнения и последствия
Если терапия не будет назначена своевременно, болезнь приводит к смерти. Среди стандартных осложнений гепатоцеребральной дистрофии можно выделить:
- Цирроз печени — возникает на последней стадии заболевания.
- Печёночная недостаточность — дисфункция печени вследствие разрушения клеток.
- Асцит — скопление жидкости в брюшной полости.
- Перитонит — воспаление брюшины.
- Варикозное расширение вен — возникает из-за повышенного давления в сосудах.
- Расширение вен пищевода, вследствие чего возникает внутреннее кровотечение. Его можно определить по таким признакам: рвота с кровью, кал окрашивается в неестественный тёмный цвет, артериальное давление снижается, сердечные сокращения учащаются.
- Нервно — психический синдром — проявляется спутанностью сознания, возникают поведенческие расстройства, нервно-мышечные патологии.
- Опухоль печени, образуется из-за систематического повреждения. Новообразование очень быстро прогрессирует и практически не поддаётся лечению.
- Почечная недостаточность — наступает по причине нарушения фильтрации в крови отравляющих веществ.
- Печёночно-лёгочный синдром — снижение содержания кислорода в крови, как следствие нарушения циркуляции крови в лёгких.
- Заболевания ЖКТ.
- Бесплодие, импотенция.
Симптомы патологии
Поскольку синдром Вильсона-Коновалова связан с нарушением выведения меди из организма и накоплением ее в центральной нервной системе и печени, то проявления связаны с ухудшением их функций. При болезни Вильсона-Коновалова симптомы следующие:
- Наличие вокруг радужки коричневого кольца Кайзера-Флейшнера.
- Кровоизлияния на коже и слизистых оболочках, вызванные нарушением синтеза витамина К-зависимых факторов свертывания крови в печени. Частые носовые кровотечения.
- Тугоподвижность мышц (ригидность), множественные спастические параличи и контрактуры. Иногда тонус мышц, наоборот, снижен.
- Припадки по типу эпилептических, двигательные нарушения – дрожание, непроизвольные движения мышц, судорожные плач и смех.
- Нарушение речи и глотания (см Афазия). Снижение интеллекта.
- Разрушение костей и зубов.
- Обильное потоотделение, бледная и синюшная кожа.
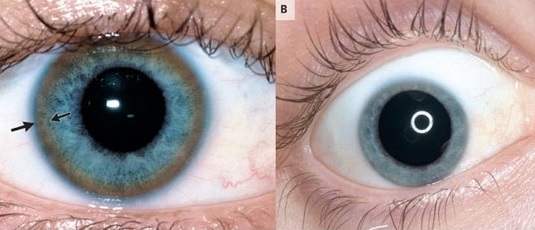 Накапливаясь в ядрах промежуточного мозга: в хвостатом теле, скорлупе, бледном шаре, черном веществе, чечевице (nucleus lentiformis), медь вызывает экстрапирамидные нарушения. Это хорея (пляска святого Витте), атетоз, дистония. Изменяется тонус мышц, в большинстве случаев они становятся ригидными, т.е. напряженными. Это сильно затрудняет движение и координацию.
Накапливаясь в ядрах промежуточного мозга: в хвостатом теле, скорлупе, бледном шаре, черном веществе, чечевице (nucleus lentiformis), медь вызывает экстрапирамидные нарушения. Это хорея (пляска святого Витте), атетоз, дистония. Изменяется тонус мышц, в большинстве случаев они становятся ригидными, т.е. напряженными. Это сильно затрудняет движение и координацию.
Хорея проявляется в беспорядочных непроизвольных жестикуляциях, которые напоминают пляску. Атетоз также является видом гиперкинеза, напоминает затянувшуюся судорогу мышц, которая прокатывается волной по телу, характерно насильственное гримасничание. При дистонии могут одновременно напрягаться мышцы-антагонисты, т.е разгибатели и сгибатели руки, например. Это причиняет больному боль.
Медь накапливается также в мозжечке и зубчатом ядре. Координация движений вследствие этого страдает (см. Нарушение координации). Накопление меди в печени вызывает повреждение сосудов и некроз (гибель) печеночных клеток – гепатоцитов. Погибшие клетки печени замещаются грубой волокнистой соединительной тканью. Медь является достаточно сильным окислителем. Повреждаются сосуды всех органов, страдают сосуды мелкого диаметра – капилляры. Это приводит к кровоизлияниям, в том числе внутри кожи. Детей постоянно беспокоят кровотечения из носа. На кожных покровах часто обнаруживают петехии и кровоподтеки.
Страдают все паренхиматозные органы, в том числе поджелудочная железа и почки. В поджелудочной железе также накапливается медь, что ведет к геморрагиям в железистую ткань. В таком случае может развиваться брюшная форма болезни Вильсона, симптомы которой включают нарушения стула, боли в животе. Цирроз печени развивается очень быстро, однако неврологические нарушения могут отсутствовать.
 Из-за стертости клинических проявлений брюшная форма болезни Вильсона-Коновалова у детей протекает молниеносно и заканчивается летальным исходом. Возможны желудочно-кишечные кровотечения вследствие повышенного давления в системе воротной вены, которая заходит в печень. Вена у входа в печень фиброзируется и воспаляется. Образуются обходные пути (шунты) между сосудами воротной и нижней полой вены, вследствие чего подкожные вены живота выбухают. Развивается выпот жидкости в брюшную полость.
Из-за стертости клинических проявлений брюшная форма болезни Вильсона-Коновалова у детей протекает молниеносно и заканчивается летальным исходом. Возможны желудочно-кишечные кровотечения вследствие повышенного давления в системе воротной вены, которая заходит в печень. Вена у входа в печень фиброзируется и воспаляется. Образуются обходные пути (шунты) между сосудами воротной и нижней полой вены, вследствие чего подкожные вены живота выбухают. Развивается выпот жидкости в брюшную полость.
Поражается при гепато-лентикулярной дегенерации и ткань почек, а именно – собирательные трубочки, проксимальные канальцы. Поэтому в моче у больных обнаруживаются глюкоза, аминокислоты, белок и соли – фосфаты, ураты. Развивается ацидоз – закисление внутренней среды.
Заболевание может протекать в дрожательной форме, когда преобладают неврологические нарушения. Начать проявляться синдром может в молодости, примерно в 20-30 лет. При этом наблюдаются психические нарушения, раздражительность. Характерны эпилептические припадки и снижение тонуса мышц тела. Мимика у таких пациентов бедная, речь монотонная. На поздней стадии заболевания возможна ригидность мышц. Для ригидной формы характерно нарушение речи и глотания. Наблюдаются контрактуры мышц.
Экстрапирамидно-корковая форма встречается реже, проявляется судорогами по типу эпилептических и заканчивается смертью лет в 6. В мозге при вскрытии видны очаги размягчения мозговой ткани, а также геморрагии и гемосидерин – продукт распада гемоглобина.
Терапия патологии
Болезнь Вильсона сопровождается увеличением количества меди в кровотоке. Лечение разрабатывается таким образом, чтобы снизить ее концентрацию. Пациентам показано придерживаться диетического питания, которое заключается в исключении богатых медью продуктов. Пациентам не рекомендуется употребление изделий из какао и шоколада. Они должны отказаться от грибов и печени. Употребление любых видов орехов специалисты запрещают.
В период протекания болезни рекомендовано применение медикаментозной терапии. Наиболее часто больным делают назначение Пеницилламина. Суточная дозировка препарата составляет 1,2-2 грамма. Она определяется доктором в соответствии с индивидуальными особенностями пациента и степенью тяжести протекания патологии. При регулярном применении лекарства можно добиться стойкого клинического улучшения и даже полного устранения симптоматики. Параллельно рекомендовано принимать препараты, в состав которых входит большое количество витамина В6.
Лечение Пеницилламином требует придерживаться специальной схемы. На начальных этапах больному назначают 150 миллиграмм препарата через день. Такой схемы пациент должен придерживаться 7 дней. Следующую неделю лекарство принимается в такой же дозировке ежедневно. Далее рекомендовано каждую неделю повышать суточную дозировку медикамента на 150 миллиграмм. Максимальная доза препарата составляет 2 грамма. После улучшения состояния больного рекомендуется придерживаться поддерживающей дозировки 450-600 миллиграмм в сутки. Витамин В6 принимается в дозе 25-50 миллиграмм.
При возникновении нежелательных эффектов в виде тошноты, аллергии, болезней почек не рекомендован прием Пеницилламина. После этого прием препарата проводится в минимальной дозировке. Одновременно рекомендуется принимать 20 миллиграмм Преднизолона ежедневно. Длительность терапии заболевания составляет 10 суток. Если организм человека не принимает Пеницилламин, то рекомендовано назначение цинка Сульфата. Его необходимо принимать три раза в день по 200 миллиграмм.
Если у пациента диагностируется непереносимость Пеницилламина, то ему показан прием Унитола. Прием лекарство должен проводиться в течение месяца. После этого делают перерыв на 3 месяца. При использовании лекарства состояние человека улучшается, а симптоматика болезни стихает. Если в период протекания патологического процесса доминируют гиперкинезы, то больному рекомендовано принимать нейролептики короткими курсами. При возникновении ригидности пациентам показана:
- Леводопа;
- Тригексифенидил;
- Карбидопа.
Если консервативное лечение патологии не приносит желаемых результатов, а заболевание у пациентов протекает в тяжелой форме, то рекомендовано проведение хирургического вмешательства. В современных зарубежных клиниках проводятся операции по пересадке печени. Если исход хирургии положителен, то это приводит к улучшению состояния пациента и восстановлению процесса обмена меди в организме. В дальнейшем для терапии патологического процесса рекомендовано применение иммуносупрессивной терапии. В клинической практике нашей страны все чаще применяется биогенмоперфузия. При этом методе используются изолированные живые клетки печени и селезенки. При условии правильного проведения процедуры будет гарантирована ее высокая эффективность.
Схема лечения при патологии должна назначаться только доктором, что положительно отобразится на результате.
Диета
Люди с болезнью Вильсона должны придерживаться определенной диеты, дабы не нагружать печени и не провоцировать обострение недуга.

В рацион необходимо включить:
- Много быстро и легкоусвояемых белков — например, молоко, рис.
- Необходимое количество углеводов (не должно превышать физиологическую норму) — от 250 до 580 грамм в сутки.
- Продукты с содержанием пищевых волокон — макароны, хлеб, неочищенный рис, фрукты, овощи.
- Сложные углеводы. Овощи и зелень, богатые клетчаткой, крахмалом, пектином. Например, морковь, свекла, огурцы, сельдерей, картофель, тыква, капуста и т.д.
- Фрукты и ягоды.
Необходимо снизить суточное потребление жиров. Правильное приготовление пищи, также будет способствовать улучшению состояния больного. Нужно кушать измельченную либо до готовки, либо после пищу. Можно варить, запекать, готовить на пару, но ни в коем случае не жарить.
Естественно, ни одна диета не обходится без запрещенных продуктов, и диета при БВ — не исключение.
В этой ситуации нужно исключить все продукты, содержащие медь:
- все бобовые;
- ракообразных, морепродукты;
- минеральную и любую газированную воду;
- кофе;
- курицу;
- утку, гуся;
- баранину, мясо свиньи;
- колбасу;
- печень;
- грибы;
- перец болгарский;
- щавель;
- орехи, сухофрукты;
- мёд;
- какао, шоколад.
При раннем начале терапии прогноз очень благоприятен. Главное не бояться и терпеливо соблюдать все назначения врача.
Генетика
Аутосомно-рецессивный тип наследования болезни Вильсона. 25 % вероятность рождения больного у родителей-гетерозигот
Ген болезни Вильсона — Коновалова(ATP7B) расположен в длинном плече 13-й хромосомы (13q14.3). Ген кодирует P-тип АТФазы, которая транспортирует медь в жёлчь и включает её в церулоплазмин. В 10 % случаев мутация не обнаруживается.
Хотя описано почти 300 мутаций ATP7B, в большинстве популяций болезнь Вильсона возникает в результате небольшого количества мутаций, специфичных для этих популяций. Например, для западных популяций мутация H1069Q (замена гистидина на глутамин в позиции 1069 белка) присутствует в 37-63 % случаев заболевания, в то время как в Китае эта мутация очень редка и R778L (замена аргинина на лейцин в позиции 778) встречается чаще. Относительно мало известно о влиянии мутаций на течение заболевания, хотя по данным некоторых исследований мутация H1069Q предполагает более позднее начало неврологических симптомов.
Нормальные вариации в гене PRNP могут изменить течение болезни, увеличивая возраст появления заболевания и влияя на тип симптомов, которые развиваются. Этот ген кодирует прионный белок, который активен в головном мозге и других тканях, а также, как полагают, участвует в транспорте меди.
У заболевания аутосомно-рецессивный тип наследования. То есть больной должен получить дефектный ген от обоих родителей (см. на рисунке). Люди только с одним мутантным геном называются носителями (гетерозиготы). У них могут возникать слабовыраженные нарушения метаболизма меди.
Каким образом наследуется болезнь?
Тип наследования аномального гена болезни называется аутосомно-рецессивным. Это означает, что заболеть могут только дети, получившие сразу два мутантных гена (от матери и отца). Родители являются гомозиготными носителями.
Если ребенок получает ген только от одного из родителей, то он не заболевает, но становится гетерозиготным носителем. У него возможны отклонения в обмене меди, но слабо выраженные. Насколько здорово будет третье поколение, зависит от встречи с аналогично измененной структурой хромосом жены или мужа.
Известен ген болезни — АТР7В — он обеспечивает синтез транспортирующего медь белка (церуллоплазмина). Находится на длинном плече в хромосоме №13, на участке с кодом 13q14-q21.
 Мутации связаны с заменой последовательности включения аминокислот
Мутации связаны с заменой последовательности включения аминокислот
Обнаружены разные виды мутаций у пациентов в Китае и странах Запада. Причины нарушений остаются неясными. У 10% больных вообще не выявлены генные изменения
Установлено, что в проявлении заболевания важное значение принадлежит факторам, поражающим печень. К ним относятся перенесенные инфекционные болезни (вирусный гепатит), интоксикация
Диагностические меры
Болезнь по сей день диагностируется при помощи стандартных мероприятий. С жалобами на нее нужно обращаться к гастроэнтерологу или гепатологу, так как именно печеночная форма встречается чаще всего. Дополнительно может потребоваться консультация невропатолога, офтальмолога, психотерапевта.
После осмотра нужно сдать анализ на медь, цинк и церулоплазмин (медь-содержащий белок в крови). Также рекомендуется пройти исследование мочи. Если полученных результатов будет недостаточно, дополнительно назначается биопсия печени.

Для окончательного подтверждения синдрома Вильсона надо сдать генетический анализ.